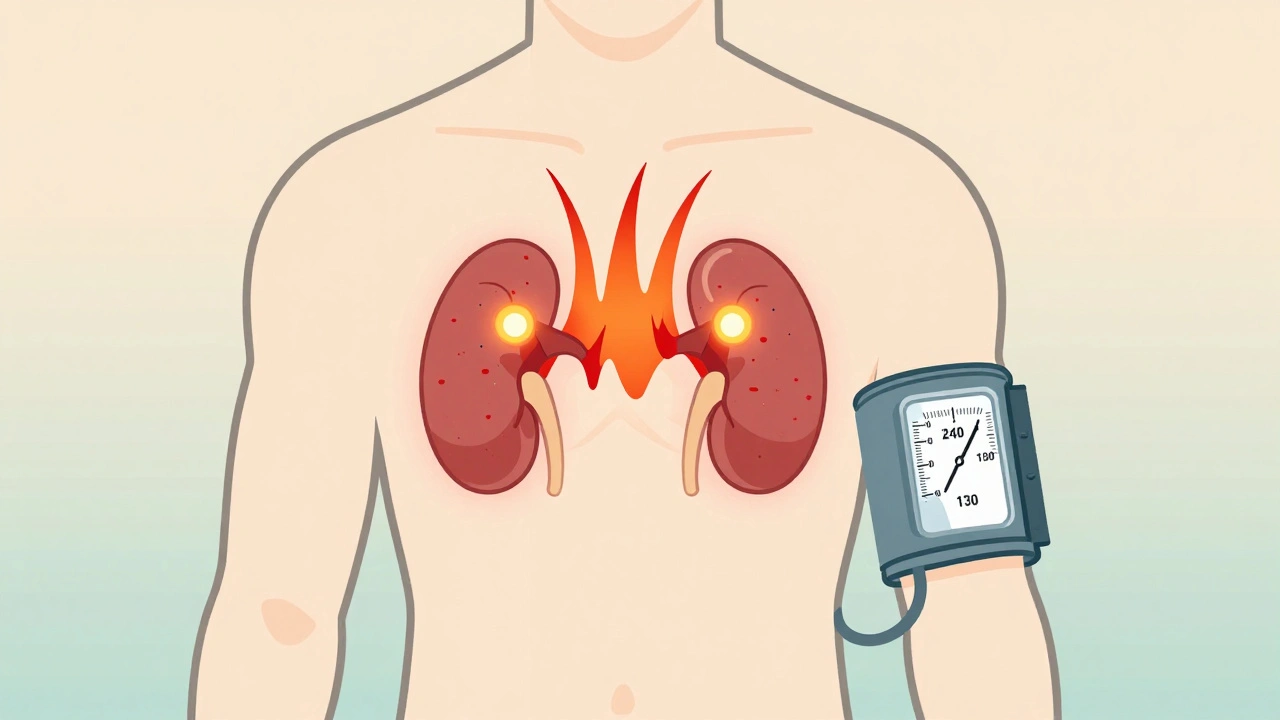
A sudden, crushing headache. Your heart pounds like it’s trying to escape your chest. You break out in cold sweat, even when you’re sitting still. Your blood pressure spikes so high it feels like your veins might burst. Then, just as quickly, it’s gone. If this has happened to you-or someone you know-more than once, it’s not just stress. It could be a pheochromocytoma, a rare but dangerous tumor hiding in your adrenal gland.
Most people with high blood pressure have what’s called essential hypertension. It’s steady, slow, and managed with lifelong pills. But pheochromocytoma is different. It’s unpredictable. It strikes in waves. And unlike most causes of high blood pressure, it can be cured-with surgery.
What Exactly Is a Pheochromocytoma?
A pheochromocytoma is a tumor that grows in the adrenal medulla, the inner part of the adrenal glands sitting on top of each kidney. These glands normally make hormones like adrenaline to help you respond to danger. But this tumor? It doesn’t know when to stop. It floods your body with catecholamines-mainly epinephrine and norepinephrine-constantly, even when there’s no threat.
It’s not cancer in most cases. About 90% of these tumors are benign. But even a benign tumor can be life-threatening because of the hormone overload. The average size is about 3 to 5 centimeters, but they can grow larger. And while most are in the adrenal glands, 10% appear elsewhere in the abdomen or chest-those are called paragangliomas.
What makes this tumor tricky is how rare it is. Only 0.1% to 0.6% of people with high blood pressure have one. That means most doctors will go their entire careers seeing just one or two cases. And because the symptoms mimic panic attacks, migraines, or even heart problems, it’s often missed.
The Classic Triad: Headache, Sweating, and Racing Heart
If you’ve ever had a panic attack, you know how scary it feels. But pheochromocytoma spells are different. They’re not triggered by stress alone. They can come from lifting something heavy, going to the bathroom, getting an injection, or even lying down.
The three most common signs are:
- Severe headaches (85-90% of patients)
- Profuse, drenching sweats (75-80%)
- Heart palpitations or a pounding pulse (70-75%)
Other symptoms include sudden paleness, nausea, anxiety that feels like a panic attack, weight loss without trying, and abdominal pain. Some people have high blood pressure that spikes above 180/110 mmHg during these episodes. Others develop low blood pressure when standing up-because the tumor messes with how your body controls circulation.
These episodes can last from a few minutes to an hour. Then, you feel fine again. That’s why many patients are told they’re just anxious. One woman in a support group waited four years and saw seven doctors before her blood pressure hit 240/130 during an ER visit-and they finally ran the right test.
How Is It Diagnosed?
There’s no single scan that catches this tumor. First, you need a blood or urine test that measures metanephrines-breakdown products of adrenaline and noradrenaline.
The gold standard is a 24-hour urine collection for fractionated metanephrines. It’s 96-99% sensitive. Blood tests for plasma-free metanephrines are almost as good, with 97% sensitivity. If your levels are more than three times the upper limit of normal, you almost certainly have a pheochromocytoma.
But here’s the catch: borderline results happen. About 15-20% of people have slightly elevated levels without a tumor. That’s why doctors don’t jump to imaging right away. If the blood or urine test is clearly positive, then they use CT or MRI to find the tumor’s location.
Advanced imaging like 68Ga-DOTATATE PET/CT is becoming more common. It’s better at spotting small tumors and those outside the adrenal glands. It’s especially useful for people with inherited forms of the disease.

Why Genetic Testing Isn’t Optional
Here’s something many people don’t realize: up to 40% of pheochromocytoma cases are inherited. That means you didn’t just get unlucky-you might carry a gene mutation passed down from a parent.
The most common genes involved are SDHB, SDHD, VHL, RET, and NF1. If you have an SDHB mutation, your risk of the tumor turning cancerous jumps to 30-50%. That’s why every single person diagnosed with this tumor should get genetic testing.
Even if no one in your family has had it, you could still carry the mutation. Studies now show that nearly a quarter of people with no family history still have a hidden genetic cause. Testing isn’t just for your sake-it’s for your children, siblings, and parents. If they carry the same gene, they can be monitored before symptoms start.
Preparation for Surgery: It’s Not Just About Cutting Out the Tumor
Surgery is the cure. But if you go into the operating room without proper preparation, you could die.
Before surgery, you need to take alpha-blockers-usually phenoxybenzamine-for at least 7 to 14 days. These drugs block the effects of excess adrenaline, preventing your blood pressure from spiking during the operation. Without them, the risk of a fatal hypertensive crisis during surgery is 30-50%.
You also need to drink more fluids and eat more salt. The tumor causes your blood vessels to stay tight for months or years, so your body has less fluid in circulation. Without volume expansion, your blood pressure can crash after the tumor is removed.
Most surgeries today are done laparoscopically-through small cuts using a camera and tools. It’s less painful, and recovery is faster. About 85% of unilateral cases are done this way. But if the tumor is very large, stuck to nearby organs, or there’s unexpected bleeding, the surgeon may need to switch to an open procedure.

What Happens After Surgery?
For most people, life changes dramatically after surgery.
Within 48 hours, blood pressure often drops to normal. Many patients stop all their blood pressure medications within a few weeks. One man on a patient forum said he was off all meds after three weeks-something he never thought possible after years of struggling with hypertension.
But not everyone has a smooth recovery.
If both adrenal glands are removed-which happens in about 10% of cases, especially with hereditary tumors-you’ll need to take lifelong steroid replacement: hydrocortisone and fludrocortisone. Without them, you can go into adrenal crisis, which can be fatal.
Some people feel exhausted for months after surgery. About 12% report chronic fatigue lasting six months or longer. That’s not just physical recovery-it’s your body relearning how to regulate itself without the tumor’s constant hormone surges.
What About Cancer?
Only about 10% of pheochromocytomas are malignant. But figuring out which ones are cancerous is hard. It’s not about how the tumor looks under a microscope. It’s about whether it spreads.
If the tumor grows into nearby tissue or spreads to lymph nodes, bones, or liver, it’s cancer. The survival rate drops sharply after metastasis. Five-year survival for metastatic cases is only about 50%.
For those cases, treatment options are limited but growing. New therapies like 177Lu-DOTATATE (a type of targeted radiation) are showing promise, with response rates around 65%. Clinical trials are also testing drugs like Belzutifan for people with VHL-related tumors.
For now, if you’ve had surgery and your tumor was malignant, you’ll need lifelong monitoring-annual MRI scans, blood tests, and check-ins with an endocrinologist.
Why This Matters More Than You Think
Pheochromocytoma is rare, but it’s one of the few causes of high blood pressure that can be completely cured. Most other forms-like kidney disease or hormonal imbalances-require lifelong treatment. This tumor? Remove it, and the high blood pressure often disappears forever.
But because it’s rare and its symptoms look like anxiety, it’s underdiagnosed. That’s why if you have unexplained, episodic high blood pressure with headaches, sweating, and palpitations, you need to ask for a metanephrine test. Don’t let it be labeled as stress. Don’t let it be ignored.
Early diagnosis saves lives. And surgery, when done right, gives people back their health.
Can pheochromocytoma be cured without surgery?
No. Surgery is the only cure. Medications like alpha-blockers can control symptoms and prepare you for surgery, but they don’t remove the tumor. Without removal, the hormone overproduction continues, and the risk of sudden, life-threatening events like stroke or heart attack remains high. Even if you feel fine for months, the tumor is still active.
Is pheochromocytoma hereditary?
Yes, in about 35-40% of cases. Mutations in genes like SDHB, SDHD, VHL, RET, and NF1 can be passed down through families. Even if you have no family history, genetic testing is recommended for everyone diagnosed. Finding a mutation means your relatives can be screened before they develop symptoms.
What happens if pheochromocytoma is missed for years?
The longer it goes untreated, the higher the risk of serious complications. Repeated spikes in blood pressure can damage your heart, kidneys, brain, and blood vessels. You’re at greater risk for heart attack, stroke, or heart failure. Some patients develop irreversible heart muscle damage. The average delay in diagnosis is over three years-many are misdiagnosed as anxiety or panic disorder during that time.
Can you live with pheochromocytoma without treatment?
Technically, yes-but it’s extremely dangerous. The tumor will keep releasing adrenaline and noradrenaline. Episodes may become more frequent or severe. Sudden spikes in blood pressure can trigger cardiac arrest, stroke, or pulmonary edema. Death can occur during anesthesia, childbirth, or even a routine medical procedure. Treatment is not optional-it’s life-saving.
What are the risks of pheochromocytoma surgery?
When done by experienced surgeons with proper preoperative preparation, the major complication rate is only about 1.2%. Risks include bleeding, infection, damage to nearby organs, and temporary low blood pressure after tumor removal. The biggest danger is not doing the prep right-without alpha-blockers, intraoperative blood pressure spikes can be fatal. Bilateral surgery requires lifelong hormone replacement.
How do I know if I should get tested for pheochromocytoma?
Get tested if you have unexplained episodes of severe headache, sweating, and rapid heartbeat-especially if your blood pressure spikes above 180/110 during these times. Also test if you have a family history of the tumor or related genetic syndromes (like neurofibromatosis or von Hippel-Lindau disease). Even if your blood pressure is normal between episodes, the test can still detect the tumor.
Comments (14)
Just had my first episode last month-headache so bad I thought I was having a stroke, drenching sweats, heart racing like I’d run a marathon. Went to three doctors who said "stress" until I insisted on metanephrines. Turns out, my levels were 5x normal. I’m scheduled for surgery next month. If you’re reading this and have similar symptoms-don’t wait. Get tested.
This is the most comprehensive breakdown of pheochromocytoma I’ve ever read. Seriously, if you’re a med student or a clinician, save this. The distinction between episodic and essential hypertension is critical, and most guidelines still overlook it.
My uncle had this and no one caught it for 5 years. He was told he had anxiety, then panic disorder, then "just old age." He ended up in the ER with a hypertensive crisis and a ruptured aorta. Surgery saved him, but he lost half his kidney function. Please, if you’re reading this and you’ve had unexplained spikes-push for the test. It’s not complicated. It’s just ignored.
Everyone’s acting like this is some miracle cure, but let’s be real-how many people even know what metanephrines are? The fact that this tumor is so often missed isn’t a failure of patients-it’s a failure of the entire medical system. You need a specialist who’s seen at least 3 cases before you get taken seriously. And guess what? Most primary docs haven’t.
Just wanted to add-my sister had this. She was 29, healthy, no family history. The genetic testing found an SDHB mutation. We all got tested. Her brother and I both carry it. We’re now on annual screening. This isn’t just about you-it’s about your whole family. Don’t wait for symptoms.
I’ve been living with this for 8 years. Every time I think I’m getting better, the episodes get worse. I’ve been on phenoxybenzamine for 6 years now. I can’t sleep through the night without wondering if tonight’s the one where my heart just gives out. I’m not ready for surgery. I’m terrified. But I’m also tired of being told I’m just "overreacting." This isn’t anxiety. It’s a ticking time bomb inside me, and no one talks about the emotional toll of living like this.
Wow. Another feel-good story about how medicine is failing people. But let’s not forget-this is a disease that affects 1 in 200,000 people. The entire healthcare system can’t be restructured just to catch this one rare condition. If you’re going to demand expensive PET scans and genetic testing for every hypertensive patient, you’re going to bankrupt the system. Priorities, people.
One must consider the ethical implications of surgical intervention in the context of autonomic dysregulation. The body’s homeostatic mechanisms are not merely biochemical-they are ontological expressions of organismic integrity. To excise a tumor without addressing the metaphysical dissonance induced by prolonged catecholamine exposure is to engage in a form of medical reductionism that ignores the holistic nature of human physiology.
Bro, I’ve been there. I had the tumor, had the surgery, now I’m on hydrocortisone 20mg twice a day. I’m not gonna lie-life after is weird. You feel like a ghost. Your body doesn’t know how to be normal anymore. But here’s the thing: you’re alive. And that’s more than most people with this get. I started a support group on Discord. DM me if you’re scared. I got you. 💪
Wait, so we’re supposed to believe that a 3cm tumor is the reason my BP spikes? What about the 20 years of processed food, caffeine, and chronic sleep deprivation? You’re blaming a tumor for the consequences of a lifestyle that’s been dumpster fire for decades. Maybe fix your diet before you ask for surgery.
For anyone reading this-don’t skip the 24-hour urine metanephrines. Plasma-free is good, but if you’re borderline, the urine is the gold standard. Also, alpha-blockers aren’t optional. I saw a case where the patient skipped prep. BP hit 280/160 in OR. Cardiac arrest. Didn’t make it. This isn’t a suggestion-it’s protocol.
Thank you for writing this. As a nurse who’s seen three cases, I can confirm-early diagnosis changes everything. I’ve watched patients go from wheelchair-bound to hiking mountains in six months. Surgery isn’t risky when done right. It’s life-giving. Please, if you suspect this, advocate for yourself. You deserve to feel normal again.
Everyone’s acting like this is a miracle, but what about the 10% who develop malignant tumors? You think surgery fixes everything? Nope. Now you’re on lifelong scans, chemo, and you’re still at risk. And don’t even get me started on the cost-$50,000 for a PET scan that insurance won’t cover. This isn’t a cure-it’s a lifetime of medical debt.
So you’re telling me I should get tested because I get headaches sometimes? That’s ridiculous. I’m 32, I drink coffee, I work 60 hours a week, I’m stressed. You think I have a tumor? Maybe try sleeping more.